

Abstract
Introduction
Recent studies utilizing NGS demonstrated that residual allelic burden at complete remission (CR) is associated with worse overall survival (OS) and relapse incidence in AML. Quantitative PCR (qPCR) based disease monitoring is current practice in CBF-AML. However, qPCR requires standardization and the result for the same sample may vary depending on several factors. Also, other known prognostic factors such as cKIT mutation require an additional test. As RNA-seq can detect gene rearrangement as well as somatic mutations, we hypothesized that RNA-seq on samples taken at diagnosis and at remission can be used to monitor these genetic alterations simultaneously and can be utilized for minimal residual disease (MRD) monitoring in CBF-AML.
Patients and Methods
This study included 42 CBF-AML patients (23 RUNX1-RUNX1T1 and 19 CBFB-MYH11 AML). Overall, 84 bone marrow samples (42 diagnosis-CR pairs) were subjected to targeted RNA-seq using Illumina TruSight Pan-Cancer panel. After read mapping, gene count was measured using HTSeq followed by DEseq2 for gene expression quantification. Average number of sequenced reads was 3.5M reads with 87% overall mapping rate. Gene fusions in diagnostic samples were detected using EricScript. All 84 samples as well as 42 samples from T-cell fraction (CD3+, as a control) were also subjected to DNA sequencing, targeting a panel of 84 genes (Agilent SureSelect custom gene panel). Average on-target coverage was 1,606x. All other computational analyses were done using R and python.
Results
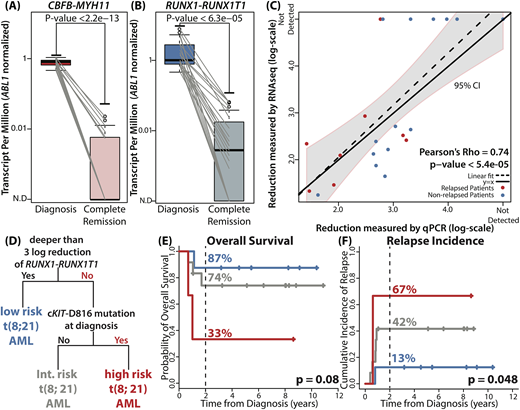
In diagnostic samples, class-defining gene fusion events were detected in all 42 patients. In CR samples, we tracked identical junctions identified in corresponding diagnostic samples. As expected, both CBFB-MYH11 and RUNX1-RUNX1T1 showed significant reduction in all CR samples compared to their corresponding diagnostic samples (p < 2.2e-13 and p < 6.3e-05, Fig A and B). CBFB-MYH11 was detectable in 6/19 CR samples (32%) and RUNX1-RUNX1T1 was detectable in 15/23 CR samples (65%). Reduction level of RUNX1-RUNX1T1 measured by RNA-seq showed positive correlation with the reduction level measured by qPCR (Pearson's Rho = 0.74, p < 5.4e-05, Fig C). As per mutational profile at diagnosis, we detected 74 mutations in 38 samples (n=38/42, 90%). NRAS (36%), KIT (36%), KRAS (17%) and, ASXL2 (17%) were commonly mutated. Survival analyses on each gene and each protein locus identified cKIT-D816 mutation as an adverse prognostic factor (HR = 3.57, [1.15 - 11.11], p = 0.028). We were able to detect all cKIT-D816 mutations in RNA-seq.
Using information from NGS, we built a prognostic model for RUNX1-RUNX1T1 AML (n = 23). Decision tree analysis identified three distinct subgroups of RUNX1-RUNX1T1 AML on the basis of reduction level of RUNX1-RUNX1T1 and mutation profile (Fig D). Consistent with previous studies, 3-log or deeper reduction of RUNX1-RUNX1T1 transcript level was the most significant prognostic factor (low risk group). The algorithm further divided the patients who failed to achieve 3-log reduction according to the presence of cKIT-D816 mutation at diagnosis (intermediate and high risk group). For three defined groups, 2-year OS rates were 87%, 74%, and 33% (p = 0.08, Fig E) and 2-year relapse incidence rates were 13%, 42%, and 67% (p = 0.048, Fig F).
Conclusion
RNA-seq can be utilized to quantify RUNX1-RUNX1T1 and CBFB-MYH11 transcripts on diagnostic and CR samples in CBF-AML. We also showed that RNA-seq can stratify RUNX1-RUNX1T1 AML patients into three risk groups according to their long-term prognosis.
No relevant conflicts of interest to declare.

